{kind=link}
{kind=link}
{kind=link}
{kind=link}
Last modified 10/26/2010.
Dr. LandryAmyloidoses are characterized by deposition of homogeneous subunits into beta-pleated sheet fibrils 75-100 angstrom in cross-section and having the properties of birefringence and selective dye affinities (Congo Red).
-and principal protein constituents
Based on X-ray fiber diffraction patterns, an antiparallel beta structure has been proposed for amyloid fibrils. Remarkably, all amyloid fibers contain a regular spacing of 4.75 angstrom, the distance between parallel strands in protein beta-pleated sheets. The model suggests that amyloid fibrils are stabilized by interstrand backbone H-bonding, as in all beta sheets, and intersheet hydrophobic and van der Waals packing interactions. Note that both of these interactions can be either intra- or inter-molecular, i.e., beta strands from separate molecules can hydrogen bond to form a beta sheet. In contrast, alpha helices form backbone hydrogen bonds only intramolecularly, between residues i and i+4 in the sequence; all intermolecular contacts involving alpha helices are mediated by sidechains and are dominated by relatively slippery hydrophobic interactions. Alpha helices tend to contribute to flexible and moveable parts of proteins, whereas beta sheets contribute rigidity. Perhaps the stability and rigidity of beta sheets makes amyloid deposits resistant to clearance.
Regardless of the detailed differences in atomic structure between native and amyloid conformations, the fact that amyloid deposition is an intermolecular process results in a concentration dependence with respect to the precursor protein monomers. Thus, the initiation and growth of amyloid fibrils depends on the rates of synthesis and degradation of the precursor protein. More precursor synthesis is associated with greater risk of amyloid disease.
One may wonder how easily an all-beta protein avoids this outcome during folding. It is likely that one job of molecular chaperones is to help proteins avoid aggregates of this kind. Numerous examples have been reported in which increased synthesis of molecular chaperones reduces intracellular protein aggregation. It is also possible that amyloidoses result from chaperone interactions gone awry
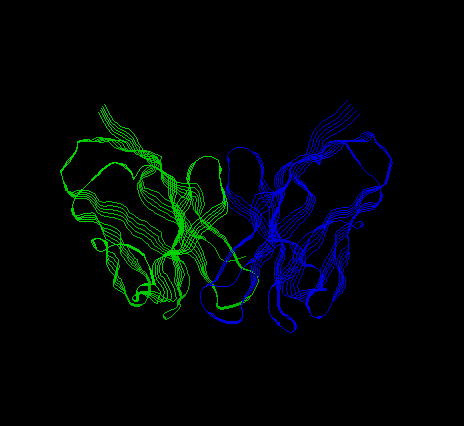
The structures of only a few amyloid-forming proteins are known, and they are rich in beta sheet. You had the opportunity to view the structure of an immunoglobulin light chain in the Fab fragment illustration for the lecture on immunoglobulin structure. Click on the choices below for somewhat less sophisticated illustrations of an immunoglobulin light chain and transthyretin.
Associated with overproduction of immunoglobulin light chain by a clonal population of B-lymphocytes (sometimes, though usually not, resulting from myeloma).
Deposits found in kidney, heart, skeletal muscle, nerves. Patients often present with kidney dysfunction.
Incidence unknown, deadly in 1-2 yr.
Associated with chronic inflammatory disease, e.g., inflammatory arthritis, granulomatous bowel disease, tuberculosis, leprosy...
Deposits found in kidney, liver, spleen. Patients often present with kidney dysfunction.
Serum amyloid A (SAA) protein produced mainly in liver. As SAA is an acute phase protein, its synthesis is induced by inflammatory cytokines. The normal function of SAA is unknown.
SAA in amyloid deposits usually has been proteolytically processed from its full length of 104 amino acids to yield a fragment composed of residues 1-76. It has been suggested that SAA amyloid forms as a result of defective or incomplete degradation of SAA.
Associated with autosomal dominant inheritance of variant transthyretin (a.k.a., prealbumin) gene. Two dozen or more variants have been identified.
Deposit location depends on variant. Nervous tissue, gastrointestinal, kidney, heart. Pathology probably arises from the mechanical distortion of tissues resulting from fiber deposition.
Incidence is rare.
The normal function of transthyretin is to transport thyroid hormones and other hydrophobic substances in the blood. The transthyretin structure may be predisposed to amyloidosis by its largely beta sheet structure. Amino acid substitutions in variant transthyretin molecules occur near one end of the molecule where they might favor intermolecular interactions. Variant transthyretins tend to be more acid labile, suggesting that amyloid formation is associated with an acidic cellular compartment, such as the lysosome, where proteolytic degradation is defective or incomplete.
Death occurs earlier in patients homozygous for the variant transthyretin gene, suggesting that amyloidogenesis is accelerated by increased protein expression or by lack of interference from normal transthyretin.
Cases may be divided into four groups as follows:
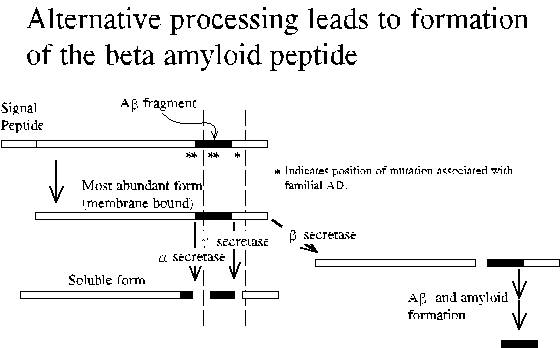
Deposits (senile plaques) in the brain are composed of an alternatively processed 40-43 residue long fragment (A-beta) of beta-PP. The disease also is characterized by the presence of neurofibrillary tangles composed largely of the microtubule-associated protein tau. Since disease is associated with mutations in beta-PP, the formation of neurofibrillary tangles is thought to be secondary to amyloid deposition. Nevertheless, it is important to note that the specific mechanism of Alzheimer's disease pathology is not known. One theory is that the deposites choke off the blood supply. Another is that A-beta is directly toxic to neurons.
Beta-PP is a cell surface receptor and secreted derivative that acts on other cells. Beta-PP physiology includes the regulated proteolytic release of soluble derivatives including hydrophobic peptides that span much of the transmembrane domain. Formation of beta amyloid involves an alternate proteolytic cleavage to yield the amino terminus of A-beta peptide and additional uncharacterized steps. A-beta peptide is actually a mixture of peptides with uniform amino-terminus and variable length, 40-43 residues. The 40-residue form is usually most abundant.
Double mutation just amino-terminal to A-beta region results in increased A-beta secretion. Disease onset occurs at 50-55 yrs.
Mutation at beta-PP residue 717 is associated with increased 42-residue A-beta. The corresponding synthetic peptide was shown to have a greater tendency to aggregate in vitro.
Results in increased beta-PP transcription leading to diffuse deposits and ultimately disease by age 60.
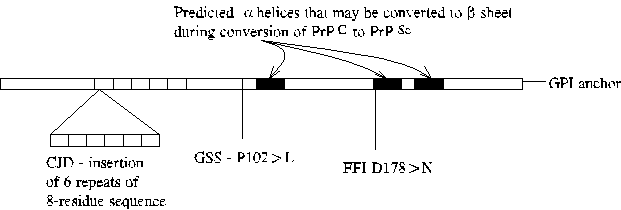
a.k.a., scrapie (sheep), bovine spongiform encephalopathy (BSE, Mad Cow Disease), kuru, Creutzfelt-Jakob Disease (CJD), Gerstmann-Straussler-Scheinker Disease (GSS), fatal familial insomnia.
Associated with autosomal dominant mutation in the prion protein precursor (PrP) or infection by infected tissue or tissue derivatives.
The long-doubted protein-only hypothesis of scrapie transmission was developed to explain (1) the failure of repeated attempts to show a nucleic acid component in the infectious agent and (2) reduced infectivity by treatments known to modify or hydrolyze proteins. The hypothetical proteinaceous infectious agent was termed the prion. In recent years, substantial evidence has shown that an essential component of the infectious agent is an alternate conformer (or possibly small aggregate) of PrP.
PrP(C) is the normal cellular isoform of the prion protein, PrP(Sc). PrP(Sc) is the principal component of amyloid plaques sometimes found in the brains of sheep infected with scrapie and in brains of humans and other animals infected with prion diseases. Conversion of PrP(C) into PrP(Sc) is thought to involve conversion of alpha-helical regions of the protein into beta sheets. Presumably, mutations associated with familial prion disease increase the likelihood of conversion. Note that different mutations result in different disease symptoms. CJD is a dementia, GSS ataxia, and FFI insomnia.
Transmission between species is characterized by low transmission rates or a long incubation time. BSE has been transmitted to mice, sheep, pigs and marmoset. Extensive studies examined the transmission between mouse and hamster. An important proof of the protein-only hypothesis was the demonstration that the incubation time for transmission from hamster to mouse was significantly shortened for a transgenic mouse bearing either hamster PrP gene or a hybrid mouse/hamster PrP gene. Prions isolated from the diseased mice were exclusively of the hamster type or hybrid mouse/hamster type, respectively. Thus, transmission is characterized by the induction of an altered form of the host gene product through its interaction with the homologous component of the infectious material.
Mice are not infected by human prions, nor are transgenic mice bearing a copy of human PrP; however, transgenic mice bearing a hybrid mouse/human PrP are infected by human prions. This suggests that an interaction between a host factor and PrP is necessary for transmission and that the mouse factor is not sufficiently similar to the human factor to interact with the human PrP. Including some mouse sequences in the otherwise human PrP restored the interaction. Recently, a prion-like phenomenon (not involving PrP) was described for yeast in which a chaperone protein was found essential for expression of the prion-like character.
End of Document