Gene Prediction
____promoter term
DNA=======================================================
RNA ~~~~~~~~~~~~~~~---------------~~~~~~~~~~~~~~~~~transcribe
RNA ~~~/~~~~~~~~~~~~|~~~~~~~~~~~~~~~\~~AAAAAA
processing
protein dsafkldfklkldfkdsdlkfprltsqwre
translation
Prediction of novel genes
Prokaryotic easier, since there is little processing to interrupt coding regions
Site
Translation coding signals
First look for long open reading frame (ORF - uninterrupted string of codons
between start and stop codons
often use a threshold value for shortest ORF you will evaluate.
Prokaryotes usually have ribosome binding site immediately upstream of initiator
AUG codon (purine rich)

Can usually identify transcriptional coding signals, too:
TATACT consensus at -10 and TTGACA at -35 from start of transcription.
Content:
codon bias - some codons preferred by given organism over others to
code for same amino acid
related to abundance of tRNAs

Where O= optimal codons, S=suboptimal, R=rare and U= unfavorable codons.
Note how few rare and unfavorable codons are used.
In contrast, the same sequence, but in a different reading (>1 above) yields
much lower quality of codon bias:

Many more rare codons and fewer optimal codons than the real reading frame.
This bias is species specific. If you look at the correct, translated reading
frame using the S. cereviseae codon bias table, the codon bias is not favorable:

Some pairs of codons tend to be found together more than others. May
be due to neighbor effects on translation. May also be due to constraints of
protein structure.
3rd base of codon (wobble) tends to fit G-C bias of rest of genome
Hidden Markov Model (HMM)
These more subtle rules may be hard to identify and quantitate, but Hidden
Markov Model programs can be used to address problem
HMM programs must be trained on known sequences to establish statistical rules
to evaluate unknowns.
So take known genes encoding known proteins to use as input into program.
Model genes will then provide statistics on codon bias, codon pairs, etc.
Depends on accuracy of model genes (poor data can obscure important rules)
and relatedness to unknown genes (i.e., need E. coli sequences to establish
rules for identifying E. coli genes)
Can try to identify coding regions and exons in genomic DNA using programs
which utilize hidden Markov models (HMM), statistical models combing information
on splice sites, codon bias, and lengths of introns and exons
Comparative methods also important for identifying genes
BLAST search can identify close or distant orthologs or paralogs
Can identify new variants of ancestral genes, or domains that have been swapped
around to make novel genes
similarity of translated reading frame to known protein is suggestive that new
reading frame is real.
Can also be used to identify genes to train HMM programs.
Eukaryotic Gene Prediction programs.
Can be based upon prokaryotic prediction programs, but require additional complexity
to reflect complexity of eukaryotic transcription, processing, and translation
most eukaryotic splicing performed by spliceosomal
complex:
splice sites determined by sequences at the ends of
introns "GTAG rule"
In many cases this requires use of neural network programs:
suites of programs that evaluate different aspects of sequences and compare
results to identify best candidates
i.e., an HMM to evaluate reading frames, AND a similarity search program to
compare to database AND a matrix program to identify consensus splice sites,
polyA sites, etc.
Some example programs
Neural network program which compares GC composition of putative gene
to flanking regions
scores splice donor and acceptor sites
evaluates ORF
scores polyA sites
Compares to EST mRNAs
Regions that score highly by one criterion are fed through other analyses (Grail-EXP
BRCA prediction benchmark)
FGENES
Pattern discrimination program
Plots functions such as exon preference vs 3' splice site score on X-Y graph.
All scores above a certain diagonal are hits
Those below are not.
FGENESH adds HMM analysis (FGENESHGC BRCA prediction
benchmark)
MZEF
Similar type of analysis to FGENES, but uses quadratic equation line to separate
winners from losers. Only tries to pick individual exons, does not try to assemble
them into a model gene.

Figure shows example of how parameter discrimination program might distinguish
"hits" from "misses". Anything above the blue diagonal would
be a hit for a linear discrimination program like FGENES, Any above the Green
arc might be hits for a quadratic parameter program like MZEF. (MZEF
BRCA prediction benchmark)
Looks for match of query DNA to model of genome composition and gene structure,
using HMM. Predicts optimal exons, but also suboptimal ones. Accuaracy of prediction
~ P value, so some useful info can be found down to P = 0.5.
Genomescan adds blastx info to increase accuracy of hits. (GENSCAN
BRCA prediction benchmark)
Benchmark of performance
In order to assess the relative strengths and weaknesses of the variious prediction
programs, a benchmark region of human genomic
DNA encoding the BRCA2
mRNA has been used as a tester sequence for all of the major prediction
programs. The results of many of these comparisons are found at the Banbury
Cross Web site. I have performed the same analysis on a 150 kb segment of
this DNA (on the reverse complement of the 13q
genomic DNA, from 3127975-2974139). and compared the results to the GenBank
accession mRNA. The results of analysis for each of the predictions described
above can be found by clicking on the respective "BRCA prediction benchmark"
link above.
Graphic
comparison of predictions using UCSC Genome Browser. Note: the nucleotide
position numbers have changed with the revised draft of the genome.
Similar
view with ensemble browser.
Public Human Genome project:
Used Ensemble and Genie programs
Ensemble uses prediction of Genscan (an HMM program), then checks these predictions
against ESTs, mRNAs and protein motifs in known databases
Merged these predictions (35,500) with those from Genie, which tries to match
5' end ESTs with 3' end ESTs to make full-length predictions
Also merged with known sequences from RefSeq
Came up with total of 31778 predicted
14,882 from known genes
12,839 from Ensemble
4,057 from Ensemble-Genie
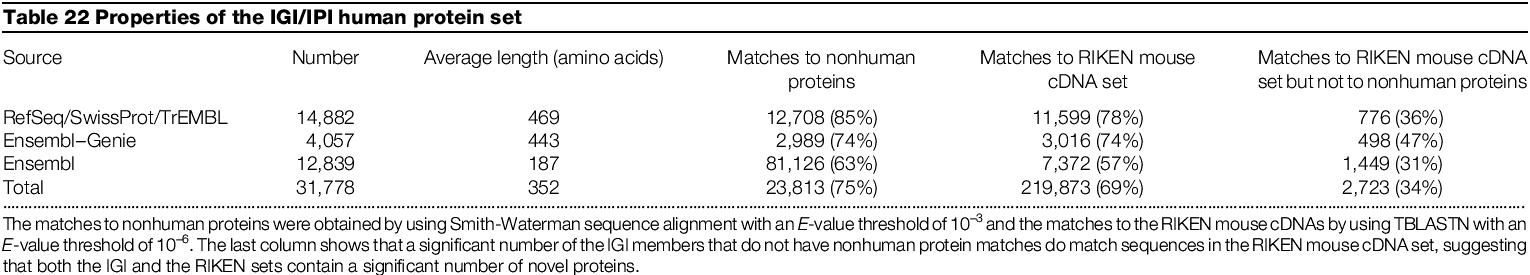
Matches to mouse cDNAs which do not match known proteins may indicate proteins
of novel function previously not identified
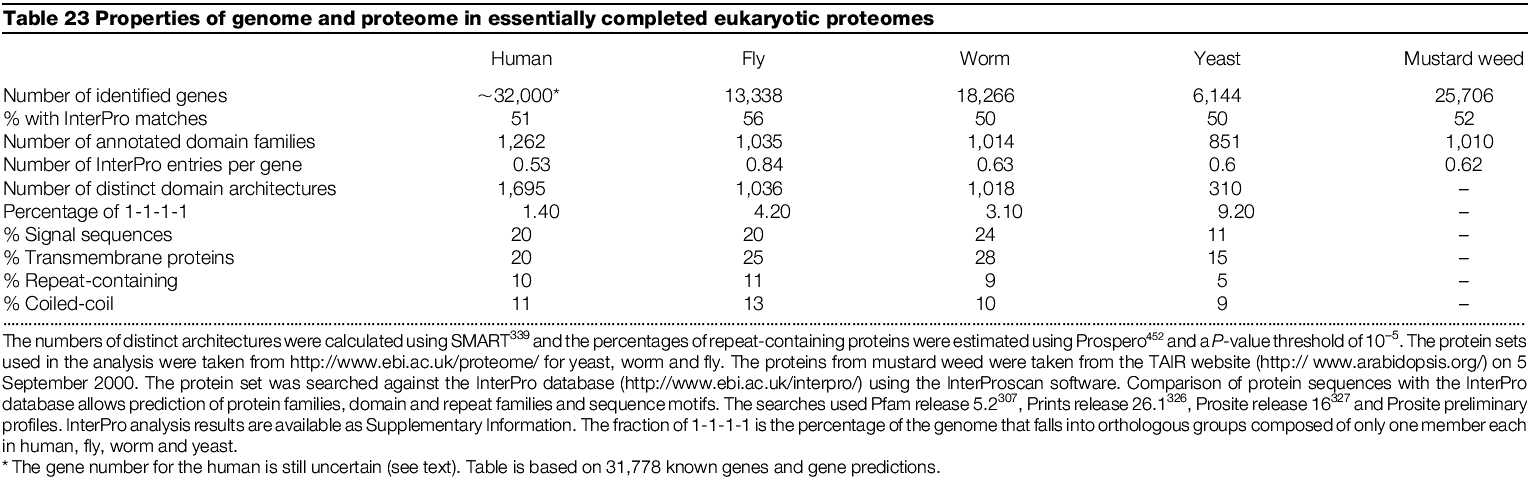
Comparison of protein coding capacity of sequenced genomes (proteomes)
GBCH723 Home Page

