Chemotherapy is the primary means of treating protozoan infections. Successful chemotherapy depends in a large part on the ability to exploit metabolic differences between the pathogen and the host. A problem confronting chemotherapy is the ability of the pathogen to mutate and become drug resistance. Specific examples, of mechanisms of drug action and resistance are discussed below.
|
Selective Toxicity
|
- unique target in parasite
- discrimination between host and parasite targets
- target more important to parasite than host
- greater drug accumulation by parasite
- drug activation by parasite
|
Drugs act by specifically interferring with cellular or biochemical processes, often called 'targets'. The classic example of a drug target is an enzyme which is inhibited by the drug. Effective drugs will exhibit a selective toxicity for the pathogen as compared to the host. Many factors contribute to this selective toxicity (Box) and these factors are not mutually exclusive. Rational drug design seeks to exploit these various factors to develop drugs which are highly toxic to the pathogen and at the same time exhibit minimal toxicity to the host.
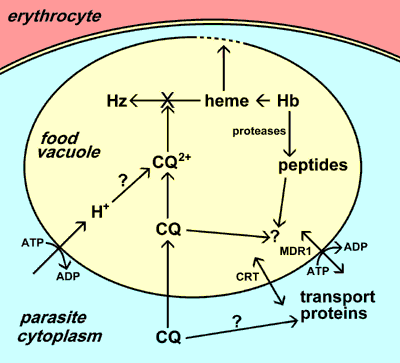
The food vacuole is a lysosome-like organelle in which the breakdown of hemoglobin and the detoxification of heme occurs (see a more detailed discussion of the food vacuole). Chloroquine concentrates up to several 1000-fold in the food vacuole of the parasite. Possible mechanisms for this selective accumulation of chloroquine in the food vacuole are: 1) protonation and ion trapping of the chloroquine due to the low pH of the food vacuole; 2) active uptake of chloroquine by a parasite transporter(s); and/or 3) binding of chloroquine to a specific receptor in the food vacuole.
 |
|
Chloroquine (CQ) accumulates in the food vacuole of the parasite. This accumulation may involve ion trapping following protonation, specific transport, and/or binding to a receptor (eg., heme). The major action of chloroquine is to inhibit the formation of hemozoin (Hz) from the heme released by the digestion of hemoglobin (Hb). The free heme then lyses membranes and leads to parasite death. Chloroquine resistance is due to a decreased accumulation of chloroquine in the food vacuole. Two different transporters (CRT and MDR1) have been implicated in resistance. The functions of these transporters and their exact roles in chloroquine resistance are not known.
|
The exact contributions of these three postulated mechanisms is not clear, but it is generally accepted that chloroquine exerts it toxic effect by interferring with the conversion of free heme to hemozoin. Large quantities of heme are released as a result of hemoglobin digestion in the food vacuole. The free heme can lyse membranes, lead to the generation of reactive oxygen intermediates, and inhibit many other processes and thus is quite toxic. Heme is detoxified in the food vacuole via a biocrystallization process in which the heme is sequestered into large insoluble crystals called hemozoin or the malarial pigment. [See more detailed description of hemozoin formation.] The exact mechanism by which chloroquine inhibits hemozoin formation is not known, but chloroquine can bind heme and this binding may prevent the heme from being incorporated into the hemozin crystal. Parasite killing is therefore a result of the accumulation of metabolic wastes (ie, heme) associated with the digestion of hemoglobin.
Other quinoline containing anti-malarials, such as mefloquine and quinine, also appear to affect the food vacuole. However, it in not clear whether these drugs bind heme or affect the formation of hemozoin. Furthermore, these drugs are weaker bases than choroquine and may not exhibit the same degree of ion trapping within the food vacuole.
The food vacuole provides many potential drug targets. In addition to the inhibition of hemozoin formation discussed above, specific inhibitors of the proteases involved in hemoglobin digestion are also being investigated as potential antimalarials. [See more detailed discussion of food vacuole proteases.] The specialized functions of hemoglobin digestion and hemozoin formation are unique to the parasite and not found within the host. Furthermore, both functions--generation of amino acids from hemoglobin and detoxification of heme--are very important for the parasite.
Folate metabolism is the target of several antimalarials as well as drugs used against other pathogens. Reduced folates serve a co-factors in a many one-carbon transfer reactions involved in the biosynthesis of amino acids and nucleotides (see more on vitamins and co-factors). Due to its high rate of replication the malaria parasite has a high demand for nucleotides as precursors for DNA synthesis (see more on nucleotides and nucleic acids), and thus is particularly sensitive to antifolates. The two primary targets of antifolate metabolism are the de novo biosynthesis of folates and dihydrofolate reductase (DHFR).
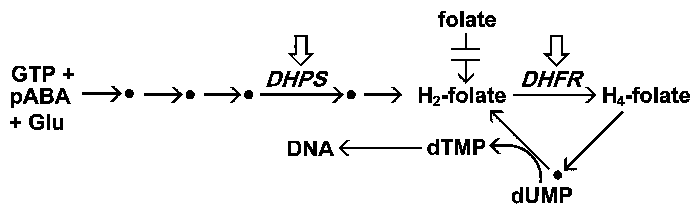
The malaria parasite synthesizes folates de novo whereas the human host must obtain preformed folates and cannot synthesize folate. The inability of the parasite to utilize exogenous folates makes folate biosynthesis a good drug target. Folate is synthesized from 3 basic building blocks, GTP, p-aminobenzoic acid (pABA), and glutamate, in a pathway involving 5 enzymes. One of these enzymes, dihydropteroate synthase (DHPS), is inhibited by sulpha-based drugs. Sulfadoxine and dapsone are two common antimalarials that target DHPS. The sulfa drugs are structural anlalogs of pABA and are converted into non-metabolizable adducts by DHPS. This leads to a depletion of the folate pool and thereby reduces the amount of thymidylate available for DNA synthesis.
 |
|
Simplified scheme of folate metabolism. The malaria parasite synthesizes folates de novo, but cannot utilize preformed folates. Folates participate as co-factors in many biosynthetic processes. Of particular note is the synthesis of thymidylate (dTMP) which is needed for DNA synthesis. The two primary targets of antimalarial drugs which target folate metabolism are denoted with the boxed arrows.
|
DHFR is an ubiquitious enzyme that participates in the recycling of folates by reducing dihydrofolate to tetrahydofolate. The tetrahydrofolate is then oxidized back to dihydrofolate as it participates in biosynthetic reactions (eg., thymidylate synthase). Inhibiting DHFR will prevent the formation of thymidylate and lead to an arrest in DNA synthesis and subsequent parasite death. Pyrimethamine and proguanil are the two most common DHFR inhibitors used as antimalarials. These drugs inhibit DHFR from the parasite to a greater degree than the host enzyme and thus show a selective toxicity towards the parasite.
| Antifolate Combinations |
| Drugs |
Pathogens |
pyrimethamine + sulfadoxine
(or dapsone) |
Plasmodium
|
trimethoprim (or pyrimethamine)
+ sulfadiazine |
Toxoplasma
|
| trimethoprim + sulfamethoxazole |
Cyclospora, Isospora, Pneumocystis
|
|
Most often inhibitors of DHPS and DHFR are used in combination (Table) for a synergistic effect and to slow the development of drug resistance. Specific point mutations in these enzymes lead to a lower affinity for the drugs. Resistance tends to develop rapidly in the presence of drug pressure in situations where a single mutation can lead to drug resistance. The use of drug combinations will slow the development of resistance since two independent mutations must occur for resistance to develop against both drugs. Fansidar, a combination of sulfadoxine and pyrimethamine, is widely used for the treatment of uncomplicated falciparum malaria. Trimethoprim, similar to pyrimethamine, is commonly used in combination with other sulfa drugs for treatment of coccidia (Toxoplasma, Cyclospora, and Isospora) and Pneumocystis.
| Possible Redox Agents |
| Drugs |
Pathogens |
| primaquine, artemisinin derivatives |
Plasmodium
|
| metronidazole, tinidazole |
Giardia, Entamoeba, Trichomonas
|
| benznidizole, nifurtimox |
Trypanosoma cruzi
|
|
Several anti-protozoal drugs are believed to act via oxidative stress (Table). Metabolic processes will produce reactive oxygen intermediates (ROI) which can damage cellular components such as lipids, proteins, and nucleic acids (see review on oxidative stress). The high metabolic activity of most protozoan pathogens will result in production of even higher levels of ROI. This is exemplified by the malaria parasite which produces ROI as a consequence of hemoglobin digestion and the release of free heme. [See more detailed description of heme and ROI.] All cells have mechanisms by which the ROI can be detoxified (eg., redox metabolism). Drugs which specifically increase the levels of oxidative stress in the parasite may overwhelm these ROI defense mechanisms and lead to parasite death. Levels of oxidative stress can be increased by drugs that are direct oxidants, as well as by drugs which participated in oxidation-reduction cycling, sometimes called futile redox cycling.
Many of the drugs participating in redox reactions need to be activated before they are effective against their target(s). For example, metronidazole and other nitroimidazoles are broad spectrum drugs that affect a wide variety of anerobic bacteria and protozoa. These drugs are activated by a reduction of the nitro group to an anion radical. The anion radical is highly reactive and will form adjuncts with proteins and DNA leading to a loss of function. In particular, the reactions with DNA result in strand breakage and inhibition of replication and will lead to cell death. Reduction of nitroimidazoles requires strong reducing conditions and anaerobic organisms have more reduction potential than aerobic organisms. This accounts for the selectivity of these compounds for anaerobic organisms. In other words, the drugs are preferentially activated by the pathogens.
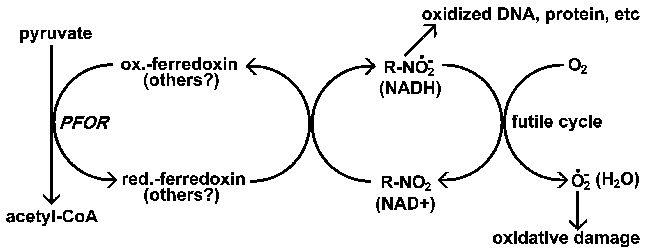
In the case of metronidazole, reduced ferredoxin appears to be the primary electron donor responsible for its reduction (Figure). There is a good correlation between the presence of the pyruvate-ferredoxin oxidoreductase (PFOR) and sensitivity to metronidazole. All three of the protozoa affected by metronidazole (Table) lack mitochondria and have PFOR similar to that found in many anaerobic bacteria. Aerobic organisms with mitochondria use pyruvate dehydrogenase instead of the PFOR for the production of acetyl-coenzyme A.
 |
|
Actions of nitroimidazoles. Nitroimidazoles (R-NO2) are activated by the parasite via a reduction to an anion radical. This highly reactive anion radical will then damage DNA and proteins resulting in parasite death. Metronidazole appears to be specifically reduced by ferredoxin in Giaridia, Entamoeba, and Trichomonas. Aerobic organisms would utilize other electron donors for the reduction of nitroimidazoles and there would also be the possibility to establish futile cycles leading to the generation of ROI in which oxygen is the final electron acceptor. Normally NAD oxidoreductases carry out redox cycling with water being the final product.
|
Nitroimidazoles (eg, benznidizole) and related nitrofuran compounds (eg, nifurtimox) also are effective against Trypanosoma cruzi. The electron donors responsible for the initial reduction of these drugs ae not known and the basis of specificity for the parasite is not clear. Both of these drugs are somewhat toxic and do not exhibit good therapeutic indices. The mechanism of nifurtimox action is believed to involve futile redox cycling following its reduction, whereas benznidizole is speculated to inhibit specific reductases and thereby decrease the ability of the parasite to remove ROI.
|
Oxidative Stress and G6PD Deficiency
Several human genetic diseases are known to confer some protection against malaria (see innate resistance). Glucose-6-phosphate dehydrogenase (G6PD) deficient individuals will have lower levels of reduced NADPH in their erythrocytes which is needed to maintain reduced glutathione. The lower levels of reduced glutathione will result in an increased sensitivity to oxidative stress since glutathione peroxidase participates in the detoxification of ROI. The increase levels of ROI due to the parasite's metabolism combined with the decrease ability G6PD-deficient erythrocytes to remove ROI will result in a premature lysis of the infected erythrocyte and therefore confer some protection against malaria. The parasite not only needs to protect itselfs against ROI but it also needs to insure that the host erythrocyte is not damaged before the parasite completes erythrocytic schizogony. In fact, it has been suggested that the parasite may supply the host erythrocyte with glutathione to increase its reducing capacity. Somewhat related, primaquine treatment is contraindicated in G6PD-deficient patients since it can cause hemolytic anemia. This is likely related to the ability of primaquine to generate ROI and the decreased reducing potential of G6PD-deficient erythrocytes.
|
|
Mechanisms of Resistance
|
- mutations in target gene
- increase production of target
- decrease drug accumulation (including increase in efflux)
- drug inactivation
|
The emergence of drug resistance severely limits the arsenal of available drugs against protozoal pathogens. Parasites have evolved numerous ways to overcome the toxicity of drugs (Box). Quite often drug resistance involves mutations in the drug target so that the drug does not bind or inhibit the target as well. Drug resistance can develop quickly in situations where a single point mutation can confer resistance. Another mechanism of drug resistance involves expressing higher levels of the target. This can be accomplished either through increased transcription and translation or gene amplification. This results in a requirement for higher levels of drugs to achieve the same level of inhibition. Decreasing drug accumulation or metabolizing the drug to non-toxic products will result in less drug reaching the target and can also contribute to drug resistance. Drug resistance can also involve the accumulation of mutations in the same or different targets which will have additive or synergistic effects. Parasites with mutations or genetic polymorphisms which confer a decrease in drug sensitivity will be selected under drug pressure.
| Proteins
and Mutations Associated with Drug Resistance |
| Protein |
Function |
Location |
Drugs Effected |
Major Mutations |
| DHPS |
folate metabolism |
cytoplasm |
sulfadoxine, dapsone |
A437G (K540E, A581G) |
| DHFR |
folate metabolism |
cytoplasm |
pyrimethamine, proguanil |
S108N (N51I, C59R, I164L) |
| CRT |
transporter |
food vacuole |
chloroquine |
K76T |
| MDR1 |
transporter |
food vacuole |
mefloquine, quinine (?) |
increased copy #, D86Y* |
| Cytochrome b |
electron transport |
mitochondria |
atovaquone |
Y268S/N/C |
| ATPase 6 |
calcium transport |
endoplasmic reticulum |
artemisinins |
S769N |
The proteins are: CRT = chloroquine resistance transporter; MDR1
= multi-drug resistance (P-glycoprotein homologue); DHFR = dihydrofolate
reductase; DHPS = dihydropterote sythetase; ATPase6 = sarco/endoplasmic
reticulum calcium-dependent ATPase orthologue. *Associated with an increased
sensitivity to mefloquine and dihydroartemisinin and a decreased sensitivity
to chloroquine. |
In some cases specific mutations have been associated
with drug resistance (Table). Fansidar (SP) resistance is correlated with specific
mutations in the enzymes targeted by sulfadoxoine and pyrimethamine (dihydropterote
sythetase and dihydrofolate reductase, respectively). Chloroquine resistance
(discuss below in more detail) has been correlated with mutations in a transporter
found on the food vacuole membrane (chloroquine resistance transporter, CRT).
Another food vacuole transporter, multi-drug resistance gene 1 (MDR1), has been
implied to play an ancillary role in resistance. The basis for resistance to
mefloquine and quinine is not clear, but the mdr1 gene has also been
implicated.
Chloroquine. Chloroquine resistance is associated with a decrease in the amount of chloroquine that accumulates in the food vacuole, the site of action for chloroquine (see above). The mechanism for this decreased accumulation is controversial. Some studies have shown that the decrease in drug accumulation is due to an increase in drug efflux. Whereas other studies suggest that diminished levels of chloroquine accumulation is more important. The observation that verapamil and related drugs can reverse the chloroquine resistant phenotype has led to speculation that an ATP dependent transporter plays a role in drug efflux and chloroquine resistance, similar to the multidrug resistance (MDR) in cancer. A MDR-like transporter, designated PfMDR1, has been identified on the food vacuole membrane. However, no definitive correlations between PfMDR1 and chloroquine resistance could be demonstrated. An ancillary role for PfMDR1 in chloroquine resistance cannot be ruled out though.
A genetic cross and mapping studies between a chloroquine resistant clone and a chloroquine sensitive clone resulted in the identification of a 36 kb region on chromosome 7 associated with chloroquine resistance. One of the 10 genes in this 36 kb region encodes a protein with 10 transmembrane domains and resembles a transporter protein similar to chloride channels. The gene has been designated as pfcrt and the protein is localized to the food vacuole membrane. Several mutations in the pfcrt gene show correlations with the chloroquine resistance phenotype and one mutation, a substitution of a threonine (T) for a lysine (K) at residue 76 (K76T) shows a perfect correlation with chloroquine resistance. Presumably these mutations affect the accumulation of chloroquine in the food vacuole, but the exact mechanism of chloroquine resistance is not known. Furthermore, the observation that chloroquine resistance has arisen relatively few times and then subsequently spread has lead to speculation that multiple genes are involved in the development of resistance (see more discussion).
Additional Reading
- Foley M and Tilley L (1998) Quinoline antimalarials.
Mechanisms of action and resistance and prospects for new agents. Pharmacology
& Therapeutics 79:55
- Hyde JE (2007) Drug-resistant
malaria -- an insight. FEBS
Journal 274, 4688-4698.
- Ouellette M (2001) Biochemical and molecular mechanisms
of drug resistance in parasites. Trop Med Int Health 6:874.
- Rosenthal PJ and Goldsmith RS (2001) Antiprotozoal
drugs. In Basic and Clinical Pharmacology, 8th edition. McGraw-Hill Companies
Inc. (On-line edition available through Stat!Ref
Books at the Tulane
Medical Library)
- Samuelson J (1999) Why metronidazole is active against
both bacteria and parasites. Antimicrob Agents Chemother. 43:1533.
- Wellems TE and Plowe CV (2001) Chloroquine-resistant
malaria. J Inf Dis 184:770.
These pages are developed and maintained by Mark F. Wiser, Tulane University. Last update May 1, 2023.