Phylogenetics is the science of estimating and analyzing evolutionary relationships. Phylogenetic relationships among micro-organisms are especially difficult to discern. Molecular biology often helps in determining genetic relationships between different organisms. Nucleic acids (DNA and RNA) and proteins are 'information molecules' in that they retain a record of an organism's evolutionary history. The approach is to compare nucleic acid or protein sequences from different organisms using computer programs and estimate the evolutionary relationships based on the degree of homology between the sequences. Nucleic acids and proteins are linear molecules made of smaller units called nucleotides and amino acids, respectively. The nucleotide or amino acid differences within a gene reflect the evolutionary distance between two organisms. In other words, closely related organisms will exhibit fewer sequence differences than distantly related organisms. In particular, the sequence of the small-subunit ribosomal RNA (rRNA) is widely used in molecular phylogeny.
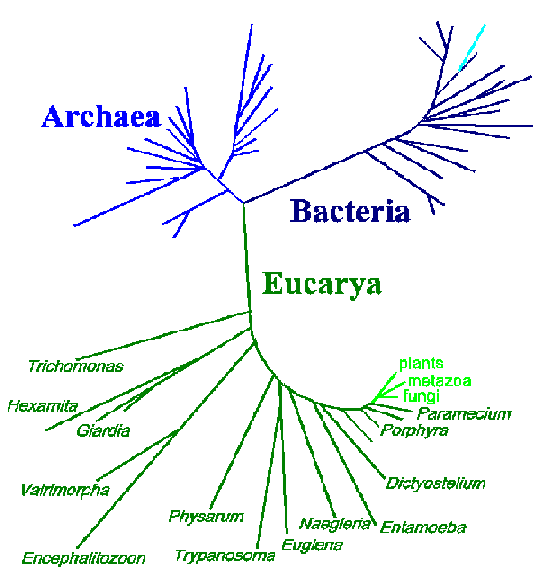
One advantage of the molecular approach in determining phylogenetic relationships over the more classical approaches, such as those based on morphology or life cycle traits, is that the differences are readily quantifiable. Sequences from different organisms can be compared and the number of differences can be established. These data are often expressed in the form of 'trees' in which the positions and lengths of the 'branches' depict the relatedness between organisms. Shown below is a three-domain tree of life based on small subunit rRNA sequences (modified from N.R. Pace, ASM News 62:464, 1996).
This tree depicts 3 major branches: eubacteria, archaebacteria, and eukaryotes. The organisms on the early branches on the eukaryote branch are all protozoa or other protists (dark green). The relative distance occupied by these organisms, as compared to the so-called higher organisms (light green), is quite notable. These data are consistent with an extremely long evolutionary history and the extreme diversity among the protozoa. However, the above tree is not entirely consistent with other criteria used to determine relationships between protozoa. Furthermore, phylogenetic trees produced from other gene sequences will produce different topologies. Possible reasons for these inconsistencies are:
The first two phenomenon result in a long-branch attraction artefact in which many slowly evolving sequences will cluster to the exclusion of a few rapidly evolving sequences. In other words, the long branches that are far apart in the lower portion of the eukaryotic branch may be a result of the experimental procedure. On the other hand, it has also been proposed that a relatively rapid (10-100 million year time span) radiation event, or 'big bang', may have occurred early in the evolution of eukaryotes giving rise to major taxa. This would also result in a similar tree topology. In addition, events like horizontal DNA transfer and gene duplications will complicate the analysis of molecular phylogenetic data.
Some of these problems are resolved by combining data into consensus trees. For example, the following tree was derived by combining protein data from elongation factor-1α, actin, α-tubulin, and β-tubulin (modified from S.L. Baldauf Am. Nat. 154, S178-188; see also Science 290, 972). This tree shows that the various groups of protozoa are quite diverse and distantly related to each other as well as showing relationships between the protozoa and other eukaryotes.
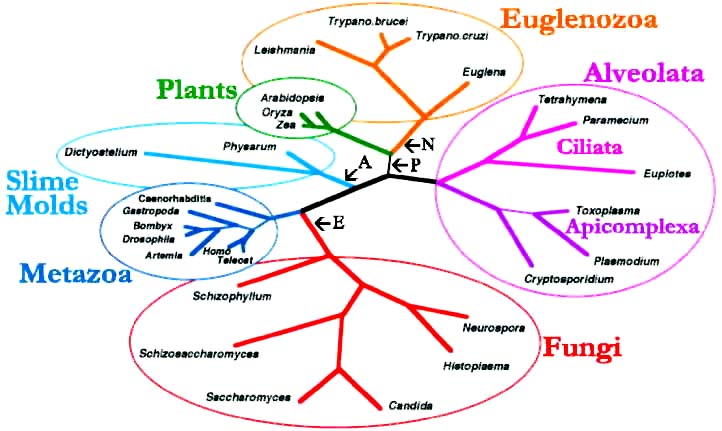 |
| The probable branch positions for some other protists are indicated by arrows (N = Naegleria; P = Porphyra, a red algae; A = Acanthamoeba; and E = Encephalitozoon, a microsporidia). |
Links back to: