Telomeres are essential for eukaryotic chromosome stability,
a fact first uncovered through the pioneering studies of Barbara McClintock.
A vast body of literature has amassed correlative evidence that support
the activation of telomerase in tumors and immortalized cell lines and
for a relationship between the telomere length and aging ([1],[2]). There
is therefore a vital need for determining the mechanism of telomere mitotic
and meiotic sizing mechanisms in context of both oncogenesis and in aging.
Recent data suggest a strong mechanistic similarity between
the yeast model system Saccharomyces cerevisiae and humans). Studies in
yeast and humans are having a synergistic relationship that is accelerating
the pace of research towards an understanding of telomere size control
and oncogenesis. Investigation of yeast TRD will therefore have a major
influence on the current view of telomere homeostasis, telomere/telomere
interactions and the stability of the terminal cap at the molecular level.
An overall understanding of the biological and medical significance of
telomere length is dependent on understanding the mechanism of telomerase
and on the means for regulating telomere size in telomerase-plus and -negative
cells. In both yeast and vertebrates, the major telomere binding protein
(Rap1 and TRF1, respectively) that associates with the double-strand tract
are central components of the telomere size machinery, in part though
a novel homeostatic counting mechanism ([3],[4],[5]).. In yeast, TRD is
superimposed on this sizing mechanism. Furthermore, TRD can act as an
efficient means for resetting telomere length in meiosis. The role of
TRD in normal human size homeostasis remains an uninvestigated area.
Telomere recombination has further significance to tumorigenesis.
Recombination mechanisms for telomere size control are present in telomerase
negative normal and oncogenic cells that lack the protein-counting machinery
[6]). Cells lacking telomerase in yeast senesce and are overtaken by cells
that rely on telomere/telomere recombination for telomere elongation.
One of these mechanisms, termed Type II recombination, is formed through
a rolling circle and/or ‘rolling loop’ mechanism, a possible
outcome of alternative means of processing of the TRD intermediate ([7],
[8]). In an analogous fashion, human cells that shorten to an extent that
precludes function terminate in cellular crisis. At a low rate, the telomeres
of immortalized cells follow an alternative recombination pathway (ALT)
that is similar to the Type II pathway. A significant proportion of sarcomas
and other tumor types use the ALT pathway of telomere elongation [6, 9].
The mechanistic relationship between elongation and recombinational deletion
in these cells remain unknown.
One exciting possibility stems from the similarity of proposed TRD intermediate to the t-loop, originally discovered as a stable complex in human and murine cells ([10]); Figure 1, top). The vertebrate t-loops are thought to function in telomere protection. T-loops have not been identified in yeast. If present, a yeast ‘t-loop’ would be expected to be transient, given the absence of an orthologue to the human TRF2, required for the stable formation of vertebrate t-loops [5]. However, lower stability structures may be present in yeast (and possibly in vertebrates) and act in telomere recombination, rather than in a capping function. Further studies on the inter-relationship between TRD and Type II recombination will lead to a clearer definition of the role(s) of t-loops in yeast and vertebrate cells ([3],[4],[5],[10])..
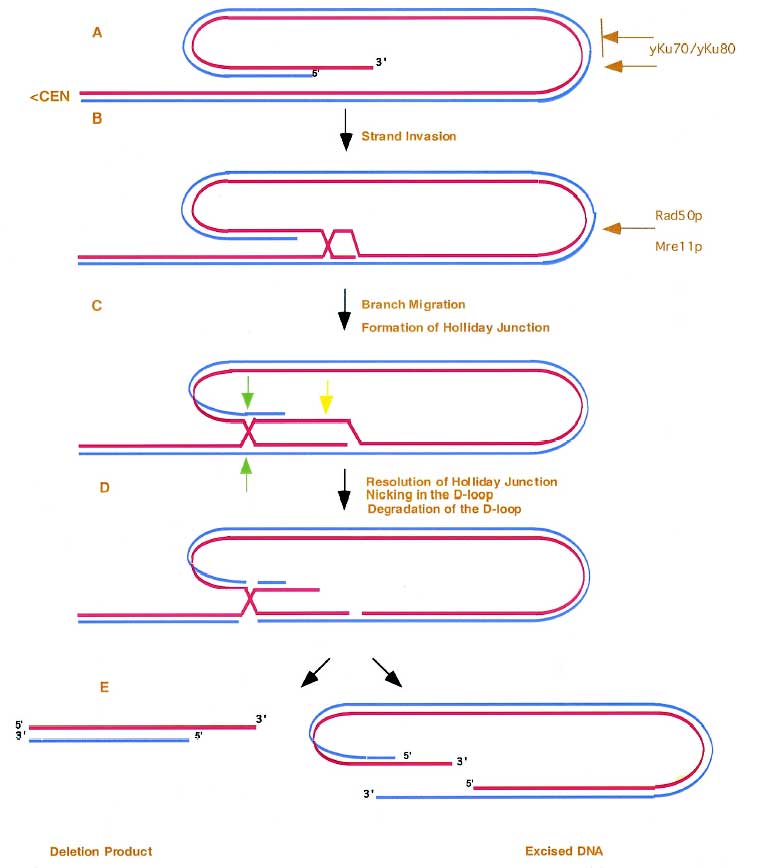
FIG. 1. Model for TRD and Type II Recombination We propose that the 3’
single strand from the telomeric terminus (A) invades distal telomere
tract sequences, leading to the formation of a t-loop-like structure After
branch migration, the displaced strand forms both a D-loop and Holliday
junction. After nicking and degrading the D-loop (yellow arrow) and resolution
of the outer strands (green arrows), both the TRD product and a linear
or circular excision product are produced (B). The TRD intermediate can
have an alternative fate if semi-conservative replication acts on the
template produced as the D-loop expands. The D-loop can then serve as
primer for lagging strand synthesis. After resolution the product will
be twice the size of the original loop. If reinvasion continues through
repetitive cycles elongated telomeres can be produced in rapid increments.
Re-invasion can occur before or after (as drawn) resolution of the Holiday
junction. Red line, the leading strand proceeding 5’ to 3’ toward
the terminus; blue line, the complementary strand. The MRX complex is
depicted as acting at the site of the t-loop junction
The significance of these studies is underscored by the recent discovery of similar catastrophic decreases in telomere size in apoptotic and pre-oncogenic cells ([11],[12]). Mre11 is essential for TRD and for alternative pathways of recombination in yeast. Importantly, Mre11 and NBS1 (Xrs2 in yeast) associates with human telomeres ([11]). Defects in the components of the MRN complex result in clinical manifestations including ataxia telangiectasia (AT)-like syndromes ([13], [14]). These syndromes are characterized by MRN DNA damage hypersensitivity, chromosomal rearrangements and the formation pre-cancerous states making the MRN and ATM pathways potential targets for medical intervention. Indeed, from a medical standpoint, an understanding of TRD and other sizing mechanisms in yeast will help us define a framework for the elucidation of the multiple recombination pathways that lead to oncogenesis in higher eukaryotic cells Our investigation of the nucleolytic and checkpoint roles of Mre11 at the telomere (e.g. in mre11A470T) is a critical part of this effort and in shifting the equilibrium between t-loop and open telomeric conformations. The ability to manipulate telomere size through TRD and checkpoint regulation can lead to a novel class of targets for the treatment of cancer.
1. Granger, M.P., Wright, W.E., and Shay, J.W. (2002). Telomerase in
cancer and aging. Crit Rev Oncol Hematol 41, 29-40.
2. Shay, J.W., and Wright, W.E. (2002). Telomerase: a target for cancer
therapeutics. Cancer Cell 2, 257-265.
3. Smogorzewska, A., van Steensel, B., Bianchi, A., Oelmann, S., Schaefer,
M.R., Schnapp, G., and de Lange, T. (2000). Control of human telomere
length by TRF1 and TRF2. Mol. Cell. Biol. 20, 1659-1668.
4. Li, B., Oestreich, S., and de Lange, T. (2000). Identification of human
Rap1: implications for telomere evolution. Cell 101, 471-483.
5. Stansel, R.M., de Lange, T., and Griffith, J.D. (2001). T-loop assembly
in vitro involves binding of TRF2 near the 3' telomeric overhang. Embo
J 20, 5532-5540.
6. Lustig, A.J. (2003). Clues to catastrophic telomere loss in mammals
from yeast telomere rapid deletion. Nat Rev Genet 9, 916-923.
7. Chen, Q., Ijpma, A., and Greider, C.W. (2001). Two survivor pathways
that allow growth in the absence of telomerase are generated by distinct
telomere recombination events. Mol Cell Biol 21, 1819-1827.
8. Bucholc, M., Park, Y., and Lustig, A.J. (2001). Intrachromatid excision
of telomeric DNA as a mechanism for telomere size control in Saccharomyces
cerevisiae. Mol Cell Biol 21, 6559-6573.
9. Reddel, R.R., and Bryan, T.M. (2003). Alternative lengthening of telomeres:
dangerous road less travelled. Lancet 361, 1840-1841.
10. Griffith, J.D., Comeau, L., Rosenfield, S., Stansel, R.M., Bianchi,
A., Moss, H., and de Lange, T. (1999). Mammalian telomeres end in a large
duplex loop. Cell 97, 503-514.
11. Ramirez, R., Carracedo, J., Jimenez, R., Canela, A., Herrera, E.,
Aljama, P., and Blasco, M.A. (2003). Massive Telomere Loss Is an Early
Event of DNA Damage-induced Apoptosis. J Biol Chem 278, 836-842.
12. Baird, D., Rowson, J., Wynford-Thomas, D., and Kipling, D. (2003).
Extensive allelic variation and ultrashort telomeres in senescent human
cells. Nature Genetics 33, 203-207.
13. Giannini, G., Ristori, E., Cerignoli, F., Rinaldi, C., Zani, M., Viel,
A., Ottini, L., Crescenzi, M., Martinotti, S., Bignami, M., Frati, L.,
Screpanti, I., and Gulino, A. (2002). Human MRE11 is inactivated in mismatch
repair-deficient cancers. EMBO Rep 3, 248-254.
14. Kang, J., Bronson, R.T., and Xu, Y. (2002). Targeted disruption of
NBS1 reveals its roles in mouse development and DNA repair. Embo J 21,
1447-1455.